
En France environ 2,6 millions de personnes prennent du Lévothyrox tous les jours. L’Agence Nationale de Sécurité du Médicament (ANSM) a demandé au laboratoire Merck, fabricant du Lévothyrox, de rendre la formule plus stable, ce qui a été fait en remplaçant notamment l’excipient par du mannitol et de l'acide citrique.
Le laboratoire a réalisé une étude de la bioéquivalence des deux formules Levothyrox, l’ancienne et la nouvelle, étude conduite dans les règles de l’art : 204 sujets sains ont reçu successivement et dans un ordre tiré au sort, la nouvelle et l’ancienne formule et des prélèvements sanguins ont été faits juste avant l’administration du traitement et ensuite 16 fois jusqu’à 72 heures après cette administration. Le taux de thyroxine dans le sérum a ainsi été mesuré 17 fois avec chaque formule pour chaque sujet.
Le laboratoire Meck a ensuite présenté cette étude sous forme de moyenne des résultats des 204 sujets, conformément aux demandes légales définies par l'ANSM et l'E.M.A.
Le scandale est là ! Ainsi que la preuve des tourments des victimes de la Nouvelle formule !
Didier Concordet et Pierre-Louis Toutain ont repris les données du laboratoire et les ont remises sous forme de tableaux et courbes, non pas des moyennes mais individualisées ! Les variations individuelles soit en hypo soit en hyper apparaissent déjà chez les sujets sains, dans des valeurs inacceptables pour des traitements à base de lévothyroxine.
Catherine Hill a repris sous forme de diapositives leur travail afin de le rendre compréhensible à des personnes non scientifiques et peut-être par les autorités sanitaires qui ne les avaient pas perçues lors de la présentation de l'analyse de bioéquivalence par Merck !
Les qualificatifs de "nocebo", de "malades hystériques" s'effondrent à l'observation des graphiques. Les variations sont telles qu'elles expliquent les malaises impressionnants dont nous avons été victimes.
Si les doses de levothyroxine s'équilibrent sous 7 jours et de manière stable quand il n'y a pas de changement de dosages, les effets et les conséquences induites par ceux-ci se sont accumulés au fil des mois où nous avons pris cette nouvelle formulation.
Il faudra une étude clinique des effets directement chez les malades pour en mettre en évidence les processus et l'ampleur de ceux-ci : dès octobre 2018, l'upgcs avait proposé l’ouverture de quelques services d’endocrinologie pour des malades sélectionnés par les associations de patients, cette proposition n’était pas inconcevable, ni matériellement impossible à financer.
France Asso Santé a fait la même proposition lors du comité de suivi du 2 juillet. ( Cf article de blog compte rendu du comité de suivi du 2 juillet).
Une étude complète et sérieuse d'un médicament doit être conduite par l'analyse de celui-ci mais aussi par un rapport sur une observation clinique à partir des patients.
Dans le cas de la Nouvelle formule, l'étude préalable d'équivalence démontrait qu'elle n'était recevable que pour 47/100 des patients !
" La différence entre les deux formules n’est inférieure à 15% que chez 47% des sujets."
Catherine Hill
C'est la première argumentation scientifique en faveur des patients depuis deux ans ! Elle nous lave des affronts "Nocebo", prouvent les effets subis et apportent un argumentaire de poids pour les actions judiciaires ! Est-ce la raison de la meilleure écoute lors du comité de suivi, possible !
Annie Notelet pour l'UPGCS
Comment la nouvelle et l'ancienne formule ont été comparées
Catherine Hill
Biostatistique et Epidémiologie
Ex Institut Gustave Roussy et
Ex membre du conseil scientifique de l’agence du médicament (AFFSAPS aujourd’hui ANSM)
Rappel de l'étude réalisée par Merck:
204 volontaires sains ont reçu chacun successivement les deux formules, dans un ordre tiré au sort.
• 102 sujets ont reçu d’abord la nouvelle formule puis, après une période de sevrage, l’ancienne formule et
• 102 sujets ont reçu d’abord l’ancienne formule puis, après une période de sevrage, la nouvelle formule.
On leur faisait 17 prélèvements par formule (donc 34 prélèvements par sujet), commençant au moment de l’administration du médicament (h0) et finissant 72 heures après (h72).
Cette bioéquivalence en moyenne est étudiée exactement comme si 204 sujets avaient pris l’ancienne formule et 204 autres sujets avaient pris la nouvelle formule.
Les patients ne sont pas intéressés par cette analyse, ils veulent connaitre la variation de la biodisponibilité entre les deux formules sujet par sujet.
Les statisticiens ne sont pas convaincus par cette analyse car ils pensent que si on fait une étude qui prend chaque sujet comme son propre témoin, il faut en tenir compte dans l’analyse.
Conclusion de cette nouvelle analyse
Un surdosage de 50% correspond au passage d’un comprimé à un comprimé et demi ; pour un médicament à fenêtre thérapeutique étroite, c’est un grand changement. Et un sous dosage de 50% correspond à une division de la dose par deux, autre grand changement.
Donc si l’expérience de ces sujets sains peut être extrapolé aux patients, on pouvait s’attendre à des surdosages et à des sous dosages importants pour une fraction non négligeable de la population.
L’agence européenne (EMA) comme l’agence française (ANSM) et probablement aussi comme l’agence des Etats-Unis (Food and Drug Administration) demande une étude de la bioéquivalence moyenne, donc Merck a fait ce qui était demandé.
Est-ce parce que c’est plus facile à obtenir ?
Pourtant les agences demandent que chaque sujet soit son propre témoin, et la FDA demande même que chaque sujet reçoive deux fois chaque formule, une moitié des sujets recevant A puis N puis A puis N et l’autre moitié recevant N puis A puis N puis A
Contestations
Certains endocrinologues qui ont défendu dans les médias et les revues médicales l’idée qu’il ne s’agissait que d’un effet « nocebo » ont tendance à rester sur cette position.
Les experts de l’ANSM font de même et discutent en plus le fait de soustraire la valeur basale (en h0), pourtant recommandé par toutes les autorités.
Merck conteste le fait de ré-analyser les données selon des règles qui ne sont pas celles des autorités.
Mais les patients ont le droit de savoir
• Et ce qui les intéresse c’est bien la bioéquivalence individuelle
• Donc merci à l’ANSM qui a fini par mettre en ligne les données de l’étude de Merck (mais pas merci pour le format inutilisable de pages photocopiées et il manque l’information sur l’ordre dans lequel les formules ont été administrées à chaque sujet)
• Et un énorme merci à Concordet,…. et Toutain qui ont retapé toutes les données dans un ordinateur, refait une analyse bien plus utile pour les patients et remis les données en ligne dans un format directement utilisable.
• Les données mises en ligne sont incomplètes dans la mesure où on ignore quels sont les sujets qui ont reçu la nouvelle formule en premier et quels sont les sujets qui l’ont eu en second. Ceci empêche de vérifier l’absence d’effet de l’ordre. Il faut obtenir cette information.
• Pourquoi avoir étudié plus de 200 sujets, alors que la FDA demande seulement au moins 24 sujets (12 par ordre) ?
• A l’heure actuelle c’est l’analyse de la bioéquivalence moyenne qui est demandée pour évaluer tous les génériques.
• Certains génériques ne sont donc probablement pas équivalents à leur médicament princeps du point de vue des patients.
Catherine Hill pour l'UPGCS



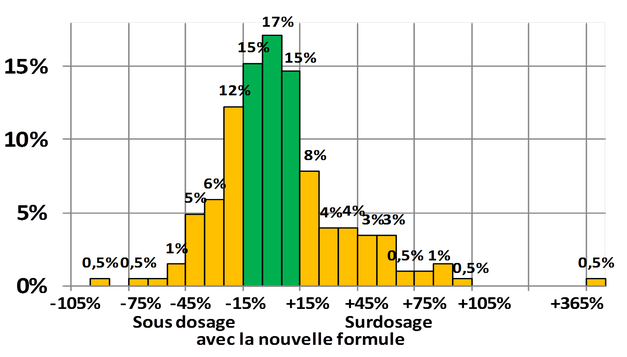
Un surdosage de 50% correspond au passage d’un comprimé à un comprimé et demi ; pour un médicament à fenêtre thérapeutique étroite, c’est un grand changement. Et un sous dosage de 50% correspond à une division de la dose par deux, autre grand changement.
Donc si l’expérience de ces sujets sains peut être extrapolé aux patients, on pouvait s’attendre à des surdosages et à des sous dosages importants pour une fraction non négligeable de la population.
Catherine HILL
Écrire commentaire
RENAUD (lundi, 08 juillet 2019 08:21)
Je remercie infiniment les personnes nommées dans cet article pour leur persévérance à nous venir en aide, à chercher ce que Merck nous cache depuis le début, l'explication de votre méthodologie afin de nous permettre à notre niveau de malade de comprendre pourquoi toutes les personnes n'ont pas réagit de la même manière car il y a un problème de bio équivalence ce qui est extrêmement important dans un traitement de la glande thyroide. Je fais malheureusement partie des personnes très touchées.
VILLACAMPA (lundi, 08 juillet 2019 12:23)
Infirmière pendant 20 ans puis inspectrice de l'action sanitaire et sociale à l'ARS de Nouvelle Aquitaine, j'ai été traitée par Lévothyrox et stabilisée depuis 1996. C'est à l'instauration de la nouvelle formule que j'ai ressenti les effets néfastes, méfiante quant à mes interprétations j'ai poursuivi avec Lévothyrox pendant près de 2 ans malgré une aggravation constante des effets secondaires dont une grande asthénie avec pour autant un bilan sanguin stable. Depuis plus d'un an je prends Eutirox de MERK en Espagne. Je revis. VILLACAMPA Frédérique
Istace lucette (jeudi, 11 juillet 2019 06:30)
Merci à vous pour vos recherches sur la vérité
Celle-ci finira par éclater.
Depuis deux ans, que de dénis et de non-dits . il est certain qu'il faut étouffer cet énorme scandale lévothyrox NF. ( nocebo, rapport flash DOOR, rapports ANSM tronqués ). Etc......
Mais la pugnacité des chercheurs, des patients et des associations face à ce scandale sanitaire vaincra.
Trop de patients ont été et sont toujours impactés par la mauvaise qualité du médicament.
Le patient a le droit de savoir...